
EPIDÉRMOLISIS BULLOSA
Resumen sobre la clasificación, repercusión y manejo de la enfermedad en el paciente
La Epidérmolisis Bullosa es un grupo raro de trastornos genéticos autosómicos, dominantes o recesivos, caracterizados por la fragilidad mecánica de los tejidos epiteliales, principalmente la piel.¹

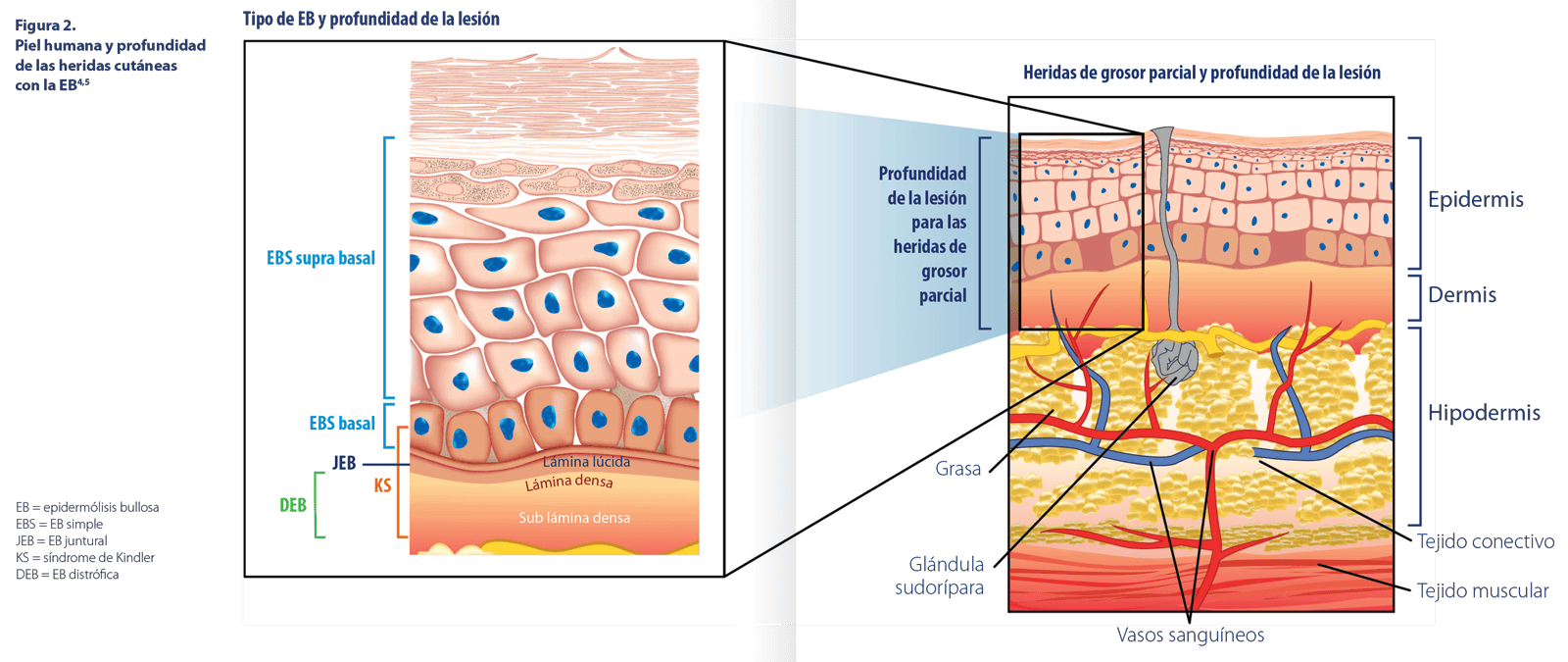
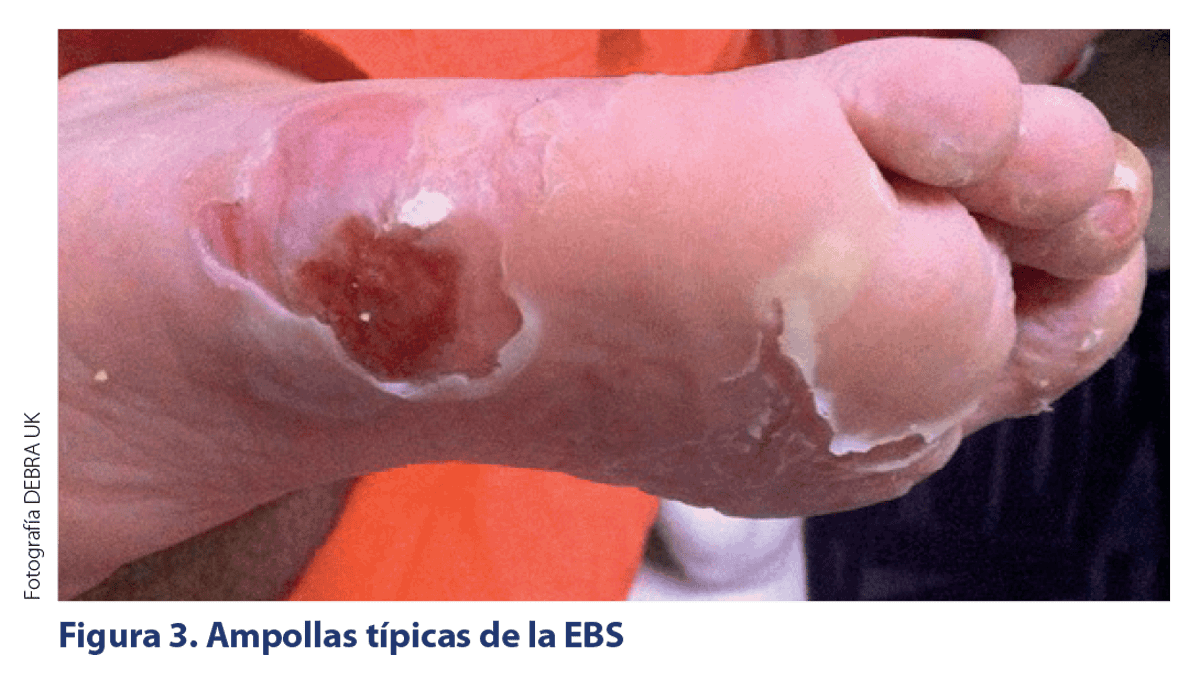
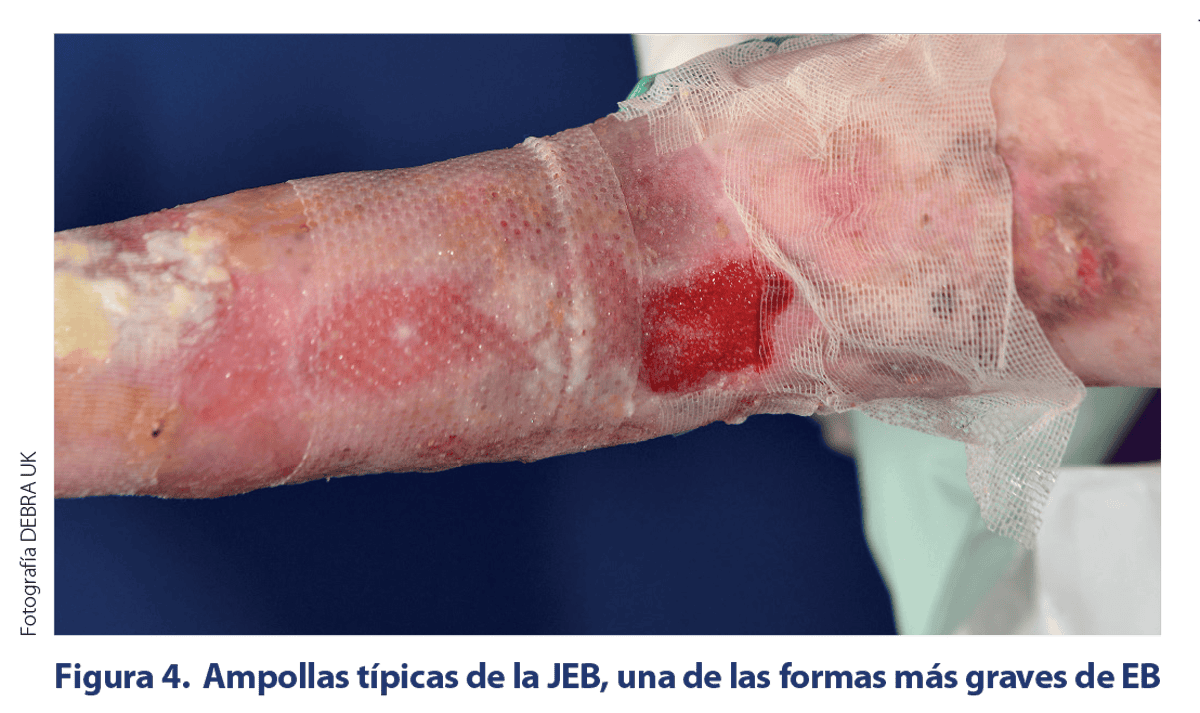
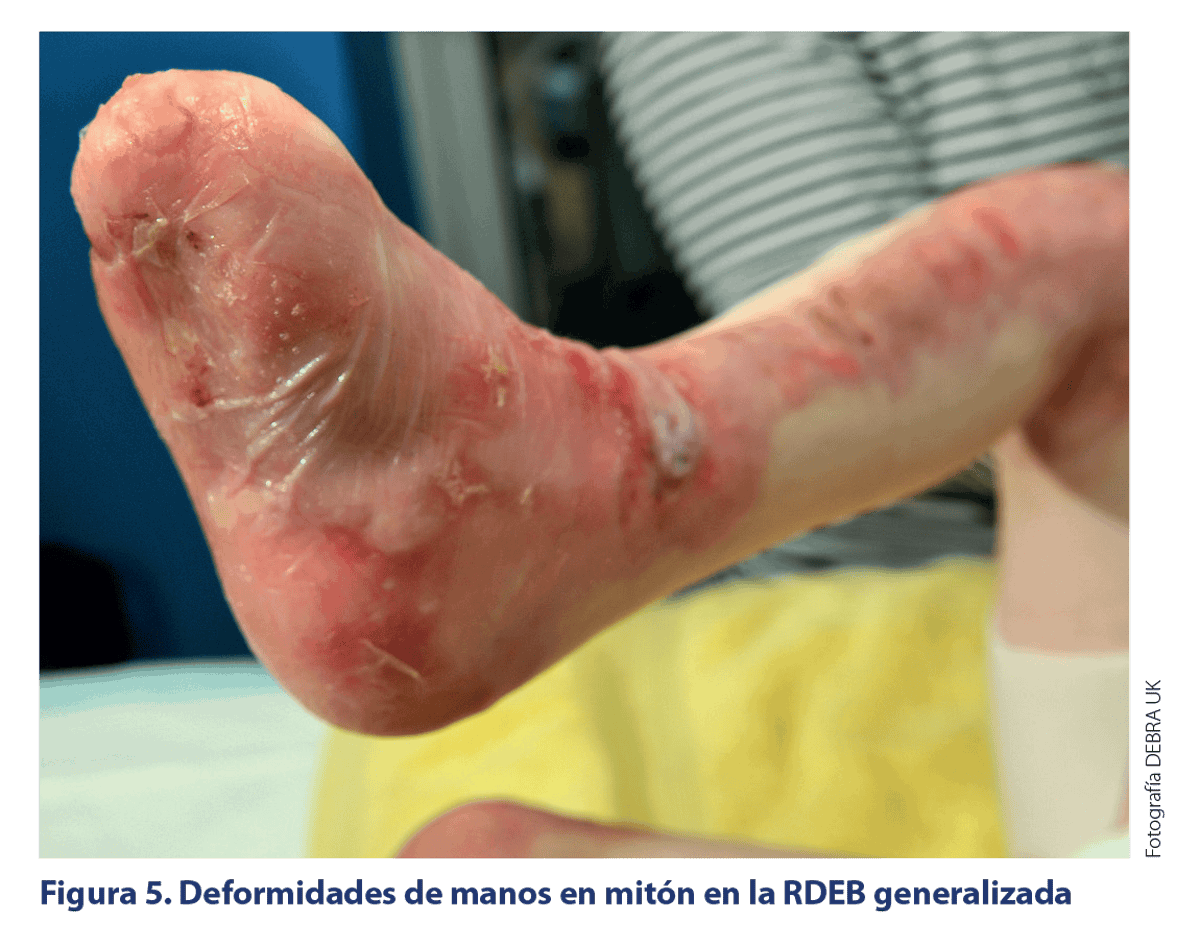
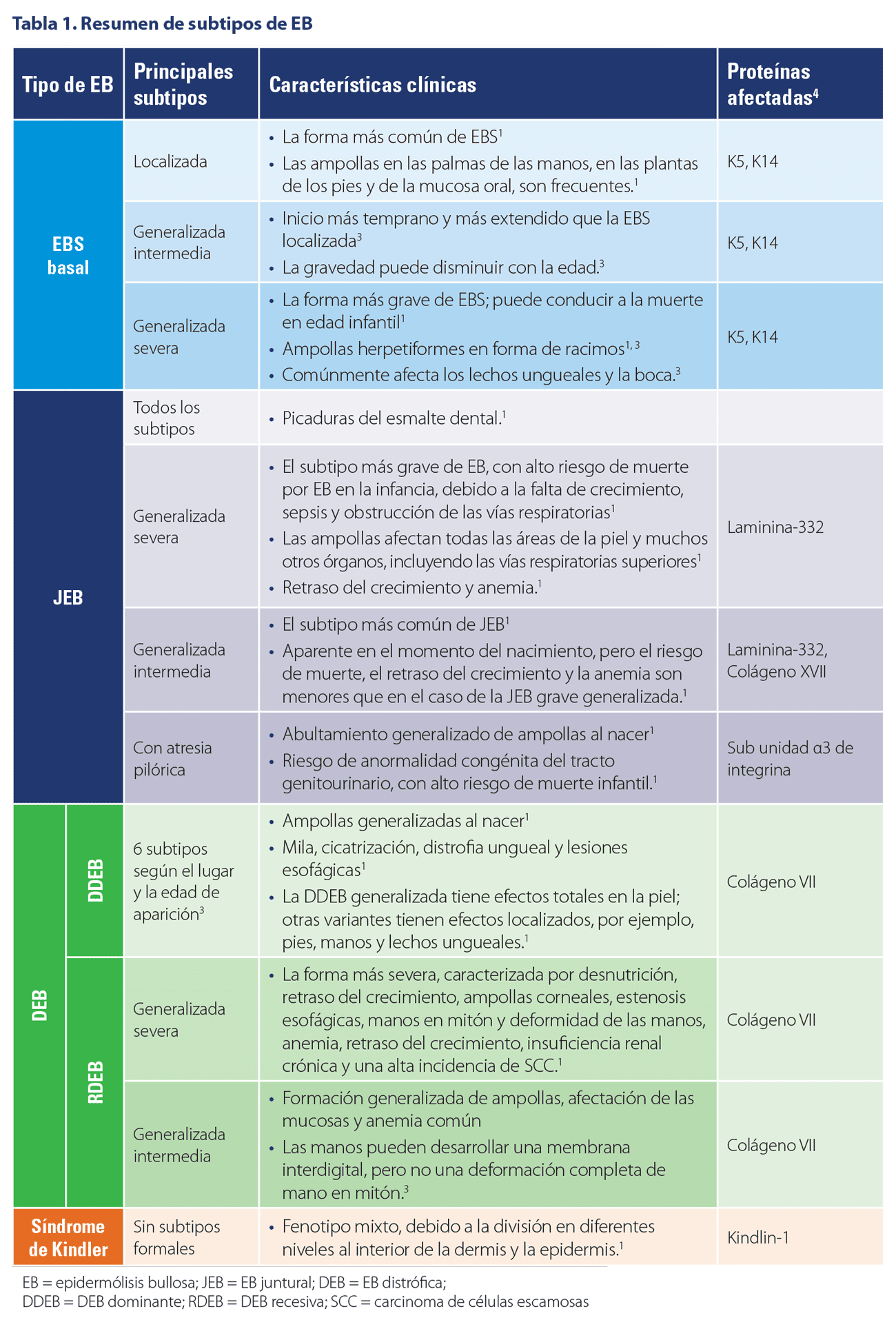

CARACTERÍSITCAS CLÍNICAS DE LOS SUBTIPOS DE EB